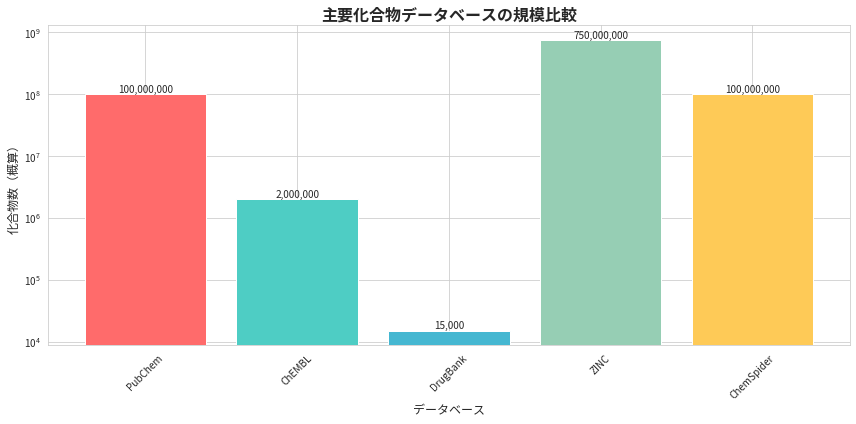
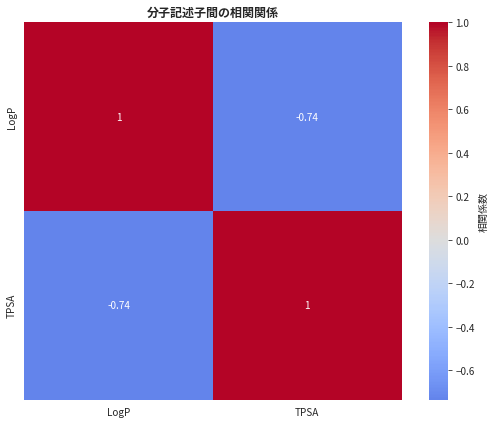
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
|
# 薬らしさ分析の可視化
if drug_like_df is not None and len(drug_like_df) > 0:
plt.rcParams['font.family'] = "Noto Sans CJK JP"
# Lipinski違反数の分布
fig, axes = plt.subplots(2, 2, figsize=(15, 12))
# 1. Lipinski違反数の分布
violation_counts = drug_like_df['Lipinski_Violations'].value_counts().sort_index()
axes[0, 0].bar(violation_counts.index, violation_counts.values, color='lightcoral', alpha=0.7)
axes[0, 0].set_title('Lipinski Rule違反数の分布', fontweight='bold')
axes[0, 0].set_xlabel('違反数')
axes[0, 0].set_ylabel('化合物数')
axes[0, 0].grid(True, alpha=0.3)
# 2. 分子量 vs LogP
colors = ['red' if not dl else 'blue' for dl in drug_like_df['Drug_Like']]
axes[0, 1].scatter(drug_like_df['Mol_Weight'], drug_like_df['LogP'], c=colors, alpha=0.7)
axes[0, 1].set_title('分子量 vs LogP (赤: 非薬物様, 青: 薬物様)', fontweight='bold')
axes[0, 1].set_xlabel('分子量 (Da)')
axes[0, 1].set_ylabel('LogP')
axes[0, 1].axhline(y=5, color='gray', linestyle='--', alpha=0.5, label='LogP > 5')
axes[0, 1].axvline(x=500, color='gray', linestyle='--', alpha=0.5, label='MW > 500')
axes[0, 1].grid(True, alpha=0.3)
axes[0, 1].legend()
# 3. 水素結合供与体・受容体数
axes[1, 0].scatter(drug_like_df['HBD'], drug_like_df['HBA'], c=colors, alpha=0.7)
axes[1, 0].set_title('水素結合供与体 vs 受容体数', fontweight='bold')
axes[1, 0].set_xlabel('水素結合供与体数')
axes[1, 0].set_ylabel('水素結合受容体数')
axes[1, 0].axhline(y=10, color='gray', linestyle='--', alpha=0.5, label='HBA > 10')
axes[1, 0].axvline(x=5, color='gray', linestyle='--', alpha=0.5, label='HBD > 5')
axes[1, 0].grid(True, alpha=0.3)
axes[1, 0].legend()
# 4. 薬物様化合物の割合
drug_like_counts = drug_like_df['Drug_Like'].value_counts()
labels = ['非薬物様', '薬物様']
colors_pie = ['lightcoral', 'lightblue']
axes[1, 1].pie(drug_like_counts.values, labels=labels, colors=colors_pie,
autopct='%1.1f%%', startangle=90)
axes[1, 1].set_title('薬物様化合物の割合', fontweight='bold')
plt.tight_layout()
plt.show()
|